理化学研究所 生命医科学研究センター 総合ゲノムミクス研究チーム チームリーダー
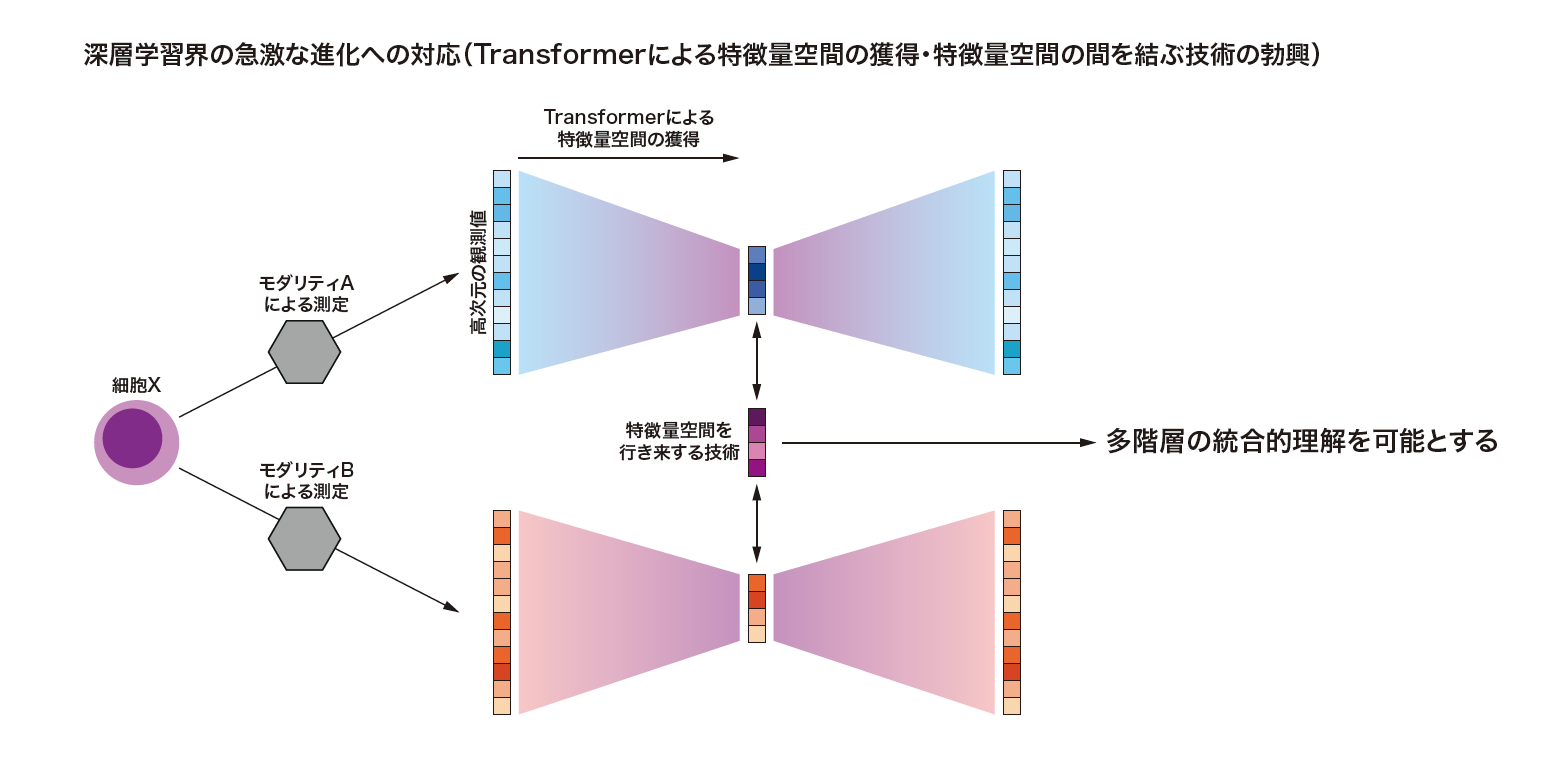
最初に、特定の「細胞X」に対して、ATAC-seqとRNA-seqの2つのモダリティ(測定方法)による同時測定を試みました。ATAC-seqは、遺伝子の転写が可能な領域(オープンクロマチン領域)を解析する手法。RNA-seq はmRNAの発現量を測定する手法です。この2種類の測定データの特徴量空間を獲得することで「細胞Xの特定の瞬間における遺伝子とタンパク質の関係」がより本質に近い姿として見えるようになると考えました。
しかし、一つの細胞に対して同時にATAC-seqとRNA-seqを行うためには、その細胞の核と細胞質を完全な形で分離しなければいけません。ところが実際は片方が高確率で損傷してしまうため、データが安定しないことがわかりました。
現在は同時測定を諦め、同じ種類の細胞を多数用意して、1つの細胞から核・細胞質のいずれかを採取し、別々に測定してデータを取得しています。細胞による個体差はありますが、データ数を増やして平均値をとることで、誤差を小さくできると考えています。
特定領域研究助成に申請した3年前は、1細胞に対する2モーダルデータを用いて、1つの特徴量空間を獲得する方法が最適解でした。
ところが言語の領域で、言語と画像の2つの特徴量空間を自由に行き来し、統合的な理解を可能にする技術が誕生しました。
医学分野でもモダリティごとに特徴量空間を獲得し、それをAIに学習させて結びつけるほうが上手くいくとわかりました。そのため現在は、1細胞に対するATAC-seqの特徴量空間と、RNA-seqの特徴量空間、さらに両方の特徴を統合的に理解できる手法の獲得を目指しています。
 言語分野では「言語と視覚のリンク」が成功し、たとえば「りんごの絵を描いて」と言語で入力すると、りんごの画像生成が可能となった
言語分野では「言語と視覚のリンク」が成功し、たとえば「りんごの絵を描いて」と言語で入力すると、りんごの画像生成が可能となった言語データに比べるとまだ極少量ですが、世界中の研究者が手分けして測定しているため、その数は日々増加しています。
先行している言語データを用いた領域では、AIが学習する教師データ数がある分量を超えると、その後は急激にAIの性能が向上することがわかりました。
医学分野でもこの現象が起きるのか、そもそもそういった特異点が存在するのかは、実際に教師データ量を増やしてみなければわかりません。どこにゴールがあるのか、まだ見えてない状態です。
 たんぱく質をコードする遺伝子数は約2万、たんぱく質の種類は約10万種類のため、言語と同規模のデータ数は必要ない。ただし、臓器における細胞の位置や時間軸なども考慮に入れると、必要なデータ数は無限に増えていく
たんぱく質をコードする遺伝子数は約2万、たんぱく質の種類は約10万種類のため、言語と同規模のデータ数は必要ない。ただし、臓器における細胞の位置や時間軸なども考慮に入れると、必要なデータ数は無限に増えていく